A Recipe for Charge Density Prediction
{kind=link}
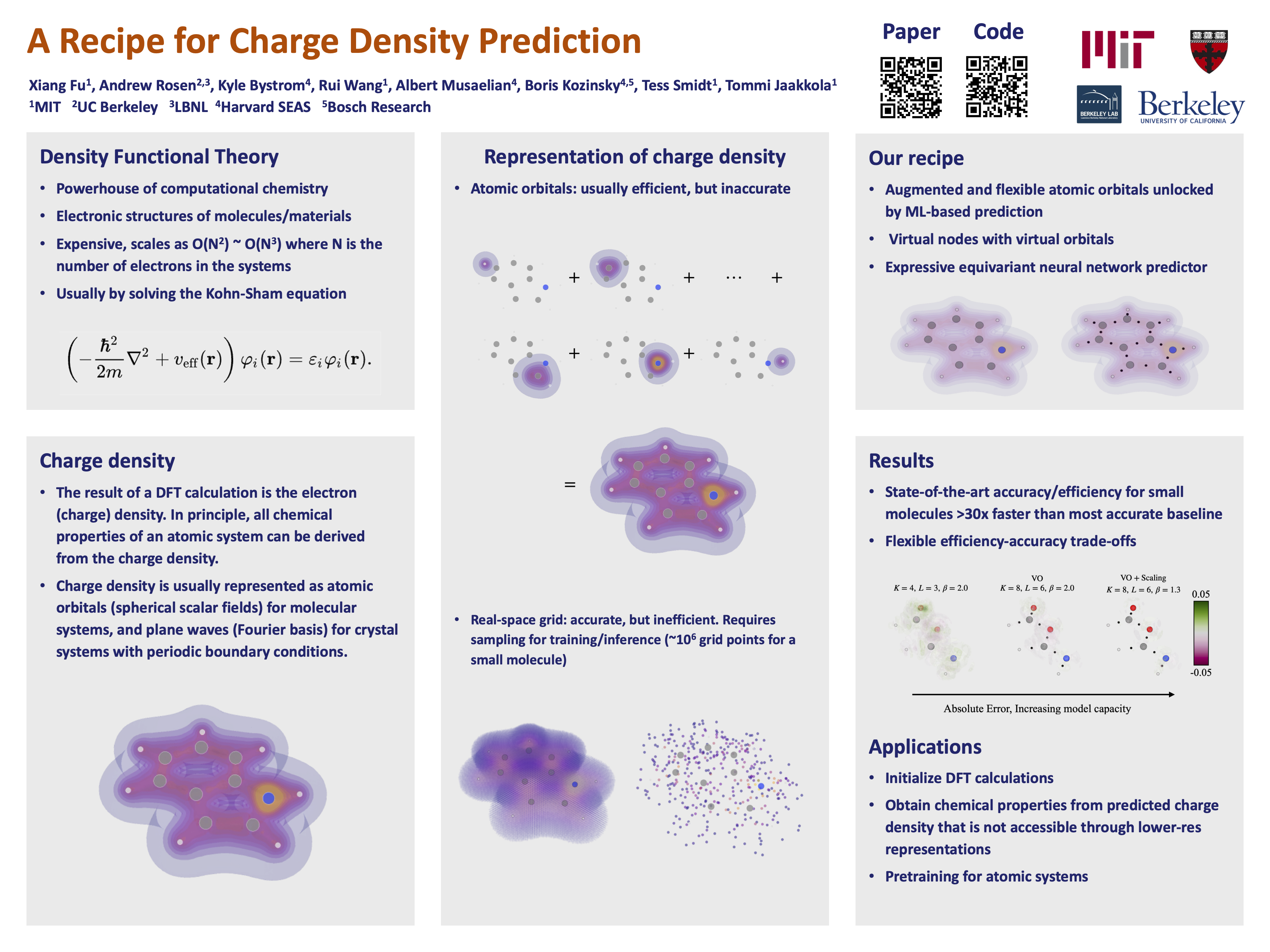
Abstract
In density functional theory, charge density is the core attribute of atomic systems from which all chemical properties can be derived. Machine learning methods are promising in significantly accelerating charge density prediction, yet existing approaches either lack accuracy or scalability. We propose a recipe that can achieve both. In particular, we identify three key ingredients: (1) representing the charge density with atomic and virtual orbitals (spherical fields centered at atom/virtual coordinates); (2) using expressive and learnable orbital basis sets (basis function for the spherical fields); and (3) using high-capacity equivariant neural network architecture. Our method achieves state-of-the-art accuracy while being more than an order of magnitude faster than existing methods. Furthermore, our method enables flexible efficiency-accuracy trade-offs by adjusting the model/basis sizes.