Graph Coloring via Neural Networks for Haplotype Assembly and Viral Quasispecies Reconstruction
{kind=link}
Abstract
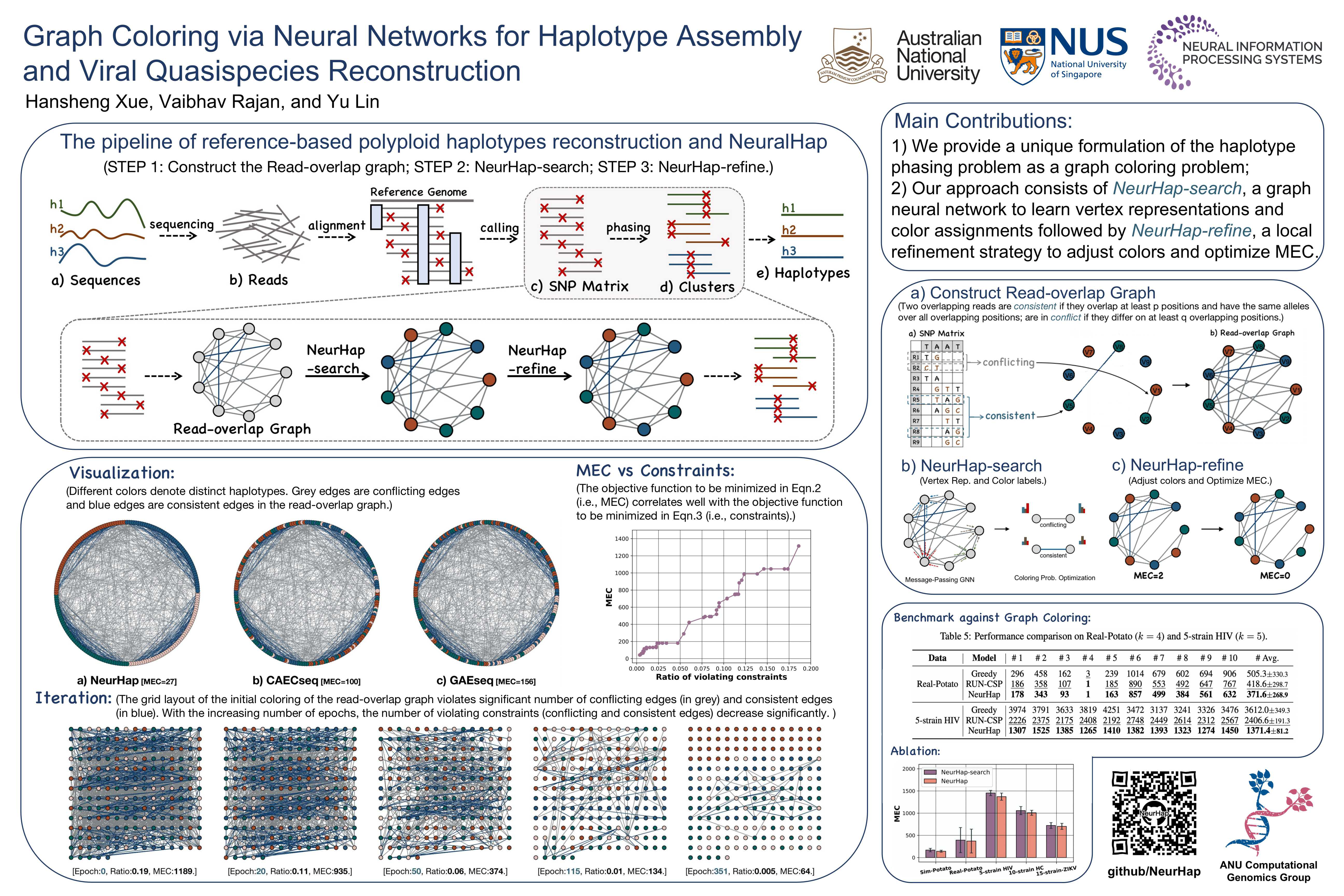
Understanding genetic variation, e.g., through mutations, in organisms is crucial to unravel their effects on the environment and human health. A fundamental characterization can be obtained by solving the haplotype assembly problem, which yields the variation across multiple copies of chromosomes. Variations among fast evolving viruses that lead to different strains (called quasispecies) are also deciphered with similar approaches. In both these cases, high-throughput sequencing technologies that provide oversampled mixtures of large noisy fragments (reads) of genomes, are used to infer constituent components (haplotypes or quasispecies). The problem is harder for polyploid species where there are more than two copies of chromosomes. State-of-the-art neural approaches to solve this NP-hard problem do not adequately model relations among the reads that are important for deconvolving the input signal. We address this problem by developing a new method, called NeurHap, that combines graph representation learning with combinatorial optimization. Our experiments demonstrate the substantially better performance of NeurHap in real and synthetic datasets compared to competing approaches.