Machine Learning for Chemical Reactions \\A Dance of Datasets and Models
{kind=link}
Abstract
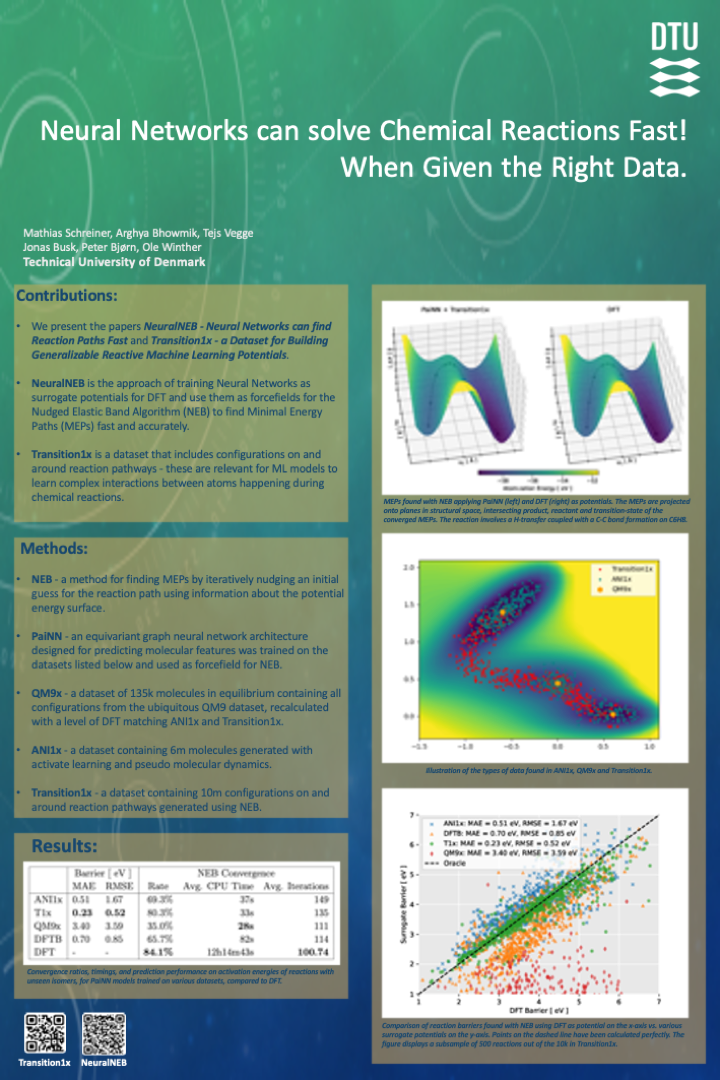
Machine Learning (ML) models have proved to be excellent emulators of Density Functional Theory (DFT) calculations for predicting features of small molecular systems. The activation energy is a defining feature of a chemical reaction, but despite the success of ML in computational chemistry, an accurate, fast, and general ML-calculator for Minimal Energy Paths (MEPs) has not yet been proposed. Here, we summarize contributions from two of our recent papers, where we apply Graph Neural Network (GNN) based models, trained on various datasets, as potentials for the Nudged Elastic Band (NEB) algorithm to speed up MEP-search. We show that relevant data from reactive regions of the Potential Energy Surface (PES) in training data is paramount to success. Hitherto popular benchmark datasets primarily contain configurations in, or close to, equilibrium, and are not adequate for the task. We propose a new dataset, Transition1x, that contains force and energy calculations for 10 million molecular configurations from on and around MEPs of 10.000 organic reactions of various types. By training GNNs on Transition1x and applying the models as PES-evaluators for NEB, we achieve a Mean Average Error (MAE) of 0.13 eV on predicted activation energies of unseen reactions, compared to DFT, while running the algorithm 1700 times faster. Transition1x is a challenging dataset containing a new type of data that may serve as a benchmark for future methods for transition-state search.